

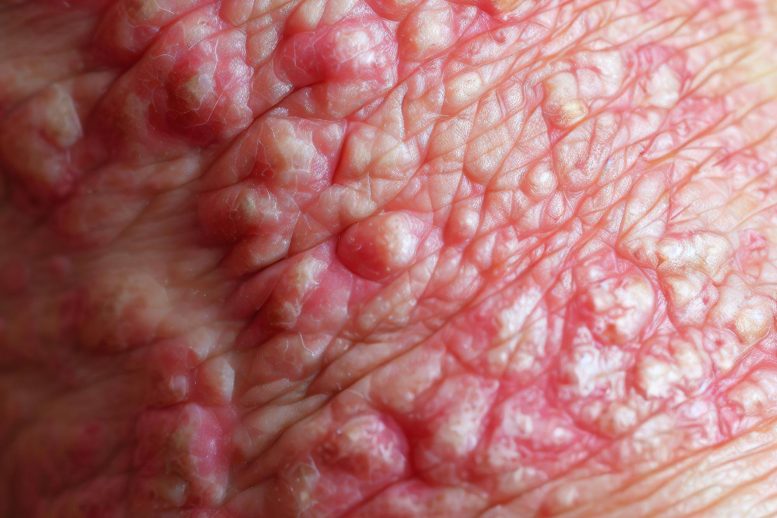
Scientists have identified genomic variants that cause disabling pansclerotic morphea, a rare inflammatory skin disorder characterized by severe skin lesions and poor wound healing, leading to deep scarring. They discovered that patients have an overactive version of the STAT4 protein, which regulates inflammation and wound healing, and found a potential treatment using a drug, ruxolitinib, that targets the inflammatory pathway. The drug, a Janus kinase (JAK) inhibitor, was observed to dramatically improve symptoms in patients.
Genome sequencing reveals genetic basis for disabling pansclerotic morphea, a severe inflammatory disease.
Researchers at the National Institutes of Health (NIH) and their colleagues have identified genomic variants that cause a rare and severe inflammatory skin disorder, known as disabling pansclerotic morphea, and have found a potential treatment. Scientists discovered that people with the disorder have an overactive version of a protein called STAT4, which regulates inflammation and wound healing. The work also identified a drug that targets an important feedback loop controlled by the STAT4 protein and significantly improves symptoms in these patients. The results were published in the New England Journal of Medicine.
The study was led by researchers at the National Human Genome Research Institute (NHGRI), part of NIH, in collaboration with researchers from the University of California, San Diego (UCSD) and the University of Pittsburgh. Researchers from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Allergy and Infectious Diseases, both part of NIH, also participated in the study.
Only a handful of patients have been diagnosed with disabling pansclerotic morphea, a disorder first described in the medical literature around 100 years ago. The disorder causes severe skin lesions and poor wound healing, leading to deep scarring of all layers of the skin and muscles. The muscles eventually harden and break down while the joints stiffen, leading to reduced mobility. Because the disorder is so rare, its genetic cause had not been identified until now.
Researchers have identified genomic variants in the STAT4 protein, causing a rare skin disorder called disabling pansclerotic morphea. A potential treatment using a JAK-inhibiting drug, ruxolitinib, has shown significant symptom improvement in patients. Credit: Ernesto Del Aguila III, NHGRI
“Researchers previously thought that this disorder was caused by the immune system attacking the skin,” said Sarah Blackstone, a predoctoral fellow within NHGRI’s Inflammatory Disease Section, a medical student at the University of South Dakota and co-first author of the study. “However, we found that this is an oversimplification, and that both skin and the immune system play an active role in disabling pansclerotic morphea.”
The researchers used genome sequencing to study four individuals with disabling pansclerotic morphea and found that all four have genomic variants in the STAT4 gene. The STAT4 gene encodes a type of protein that helps turn genes on and off, known as a transcription factor. The STAT4 protein not only plays a role in fighting infections but also controls important aspects of wound healing in the skin.
The scientists found that the STAT4 genomic variants result in an overactive STAT4 protein in these four patients, creating a positive feedback loop of inflammation and impaired wound healing that worsens over time. To stop this harmful feedback loop, they targeted another protein in the inflammatory pathway that interacts with the STAT4 molecule and is called Janus kinase, also known as JAK. When the researchers treated the patients with a JAK-inhibiting drug called ruxolitinib, the patients’ rashes and ulcers dramatically improved.
“So far, there has not been a standard treatment for this disorder because it’s so rare and not well-understood. However, our study gives an important new treatment option for these patients,” said Blackstone.
Existing treatments for disabling pansclerotic morphea are designed to halt the progression of the disorder, but previous therapies have been mostly ineffective, often with severe side effects. People with the disorder typically don’t live more than 10 years after their diagnosis.
The study suggests that ruxolitinib could be an effective treatment for patients with this disorder. Ruxolitinib is part of a broader class of drugs called JAK inhibitors, which are commonly used to treat arthritis, eczemaulcerative colitis, and other chronic inflammatory diseases.
“The findings of this study open doors for JAK inhibitors to be a potential treatment for other inflammatory skin disorders or disorders related to tissue scarring, whether it is scarring of the lungs, liver, or bone marrow,” said Dan Kastner, M.D., Ph.D., an NIH distinguished investigator, head of NHGRI’s Inflammatory Disease Section and a senior author of the paper.
“We hope to continue studying other molecules in this pathway and how they are altered in patients with disabling pansclerotic morphea and related conditions to find clues to understanding a broader array of more common diseases,” said Lori Broderick, M.D., Ph.D., a senior author of the paper and an associate professor at UCSD.
Reference: “Variant STAT4 and Response to Ruxolitinib in an Autoinflammatory Syndrome” by Hratch Baghdassarian, B.S., Sarah A. Blackstone, B.S., Owen S. Clay, M.D., Ph.D., Rachael Philips, Ph.D., Brynja Matthiasardottir, M.Sc., Michele Nehrebecky, N.P., Vivian K. Hua, B.S., Rachael McVicar, B.S., Yang Liu, Ph.D., Suzanne M. Tucker, M.D., Davide Randazzo, Ph.D., Natalie Deuitch, M.S., Sofia Rosenzweig, B.S., Adam Mark, M.S., Roman Sasik, Ph.D., Kathleen M. Fisch, Ph.D., Pallavi Pimpale Chavan, M.D., Elif Eren, Ph.D., Norman R. Watts, Ph.D., Chi A. Ma, Ph.D., Massimo Gadina, Ph.D., Daniella M. Schwartz, M.D., Anwesha Sanyal, Ph.D., Giffin Werner, B.S., David R. Murdock, M.D., Nobuyuki Horita, M.D., Ph.D., Shimul Chowdhury, Ph.D., David Dimmock, M.D., Kristen Jepsen, Ph.D., Elaine F. Remmers, Ph.D., Raphaela Goldbach-Mansky, M.D., M.H.S., William A. Gahl, M.D., Ph.D., John J. O’Shea, M.D., Joshua D. Milner, M.D., Nathan E. Lewis, Ph.D., Johanna Chang, M.D., Daniel L. Kastner, M.D., Ph.D., Kathryn Torok, M.D., Hirotsugu Oda, M.D., Ph.D., Christopher D. Putnam, Ph.D. and Lori Broderick, M.D., Ph.D., 31 May 2023, New England Journal of Medicine.
DOI: 10.1056/NEJMoa2202318




